
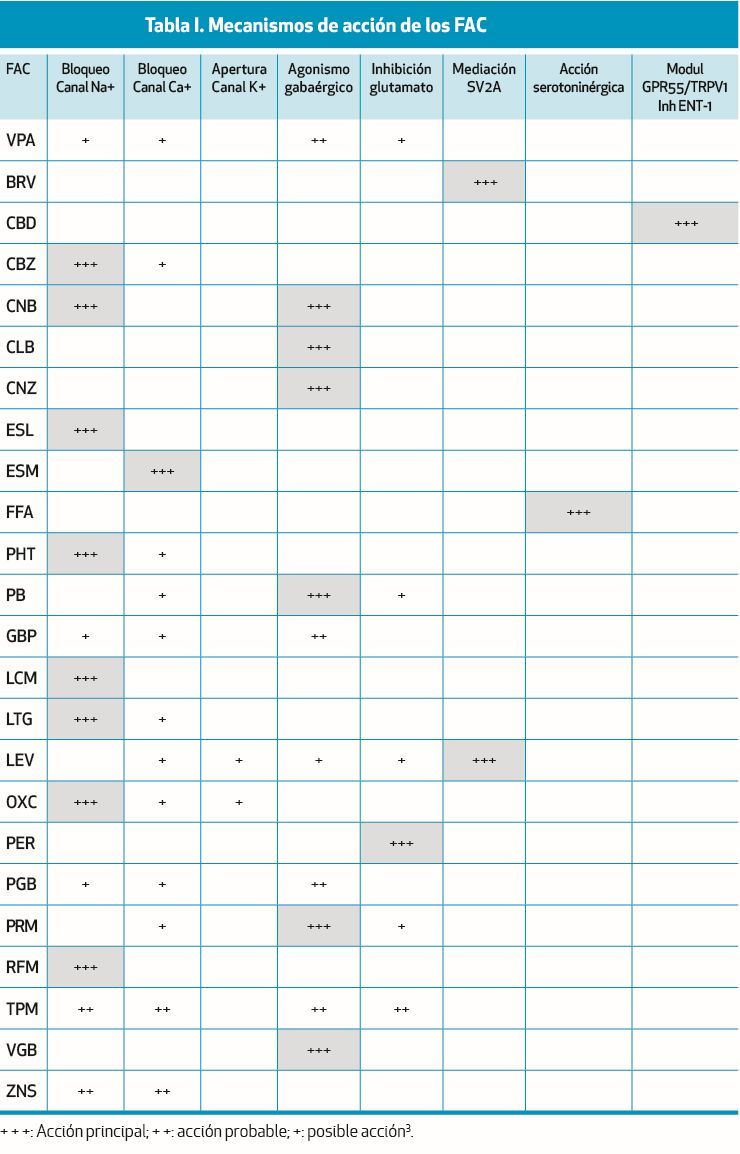
MECANISMOS DE ACCIÓN DE LOS FÁRMACOS ANTICRISIS
El efecto de los fármacos anticrisis (FAC) en la supresión de las crisis epilépticas (CE) está relacionado con la acción sobre diferentes dianas disminuyendo la excitabilidad neuronal y la hipersincronía de los circuitos cerebrales. La mayoría de los FAC presenta diferentes mecanismos de acción. La acción principal de los FAC sobre estas dianas se puede simplificar en cinco grupos1,2 (tabla I):
- Grupo 1. Acción principal sobre los canales iónicos voltajes dependientes (canales de sodio, de calcio y de potasio). Al bloquear o modular el canal de sodio se reducen las descargas neuronales repetitivas rápidas, estabilizando la membrana neuronal, disminuyendo la actividad epiléptica y la progresión de las crisis. Los canales de calcio tienen un rol crucial en el inicio y la propagación de las crisis, y la apertura de los canales de potasio facilita el restablecimiento del potencial de reposo.
- Grupo 2. Acción principal sobre el sistema GABA. El aumento de la concentración GABA o la modulación de los receptores de GABA reduce la excitabilidad neuronal.
- Grupo 3. Acción principal sobre los receptores ionotrópicos de glutamato, incluyendo los receptores AMPA, kainato, NMDA, glicina. Se pueden unir a los diferentes receptores reduciendo su actividad.
- Grupo 4 Acción principal sobre la modulación de la maquinaria que facilita la liberación sináptica de neurotransmisores. LEV y BRV se fijan a la proteína SV2A, localizada en las vesículas presinápticas facilitando la liberación de neurotransmisores inhibitorios. La GBP y la PGB se unen a la proteína α2δ, y pueden reducir la despolarización mediada por calcio reduciendo la liberación de neurotransmisores excitatorios.
- Grupo 5 Estimulación de la transmisión serotoninérgica. Fenfluoramina indicado para encefalopatías epilépticas, síndromes de Dravet y Lennox-Gastaut.
- Grupo 6. El cannabidiol, indicado para encefalopatías epilépticas, síndromes de Dravet, Lennox-Gastaut y complejo esclerosis tuberosa, con mecanismo de acción múltiple mediante un modulación de receptores GPR55 y TRPV1, además de la inhibición de adenosina por transportador ENT-1.
FARMACOCINÉTICA DE LOS FÁRMACOS ANTICRISIS
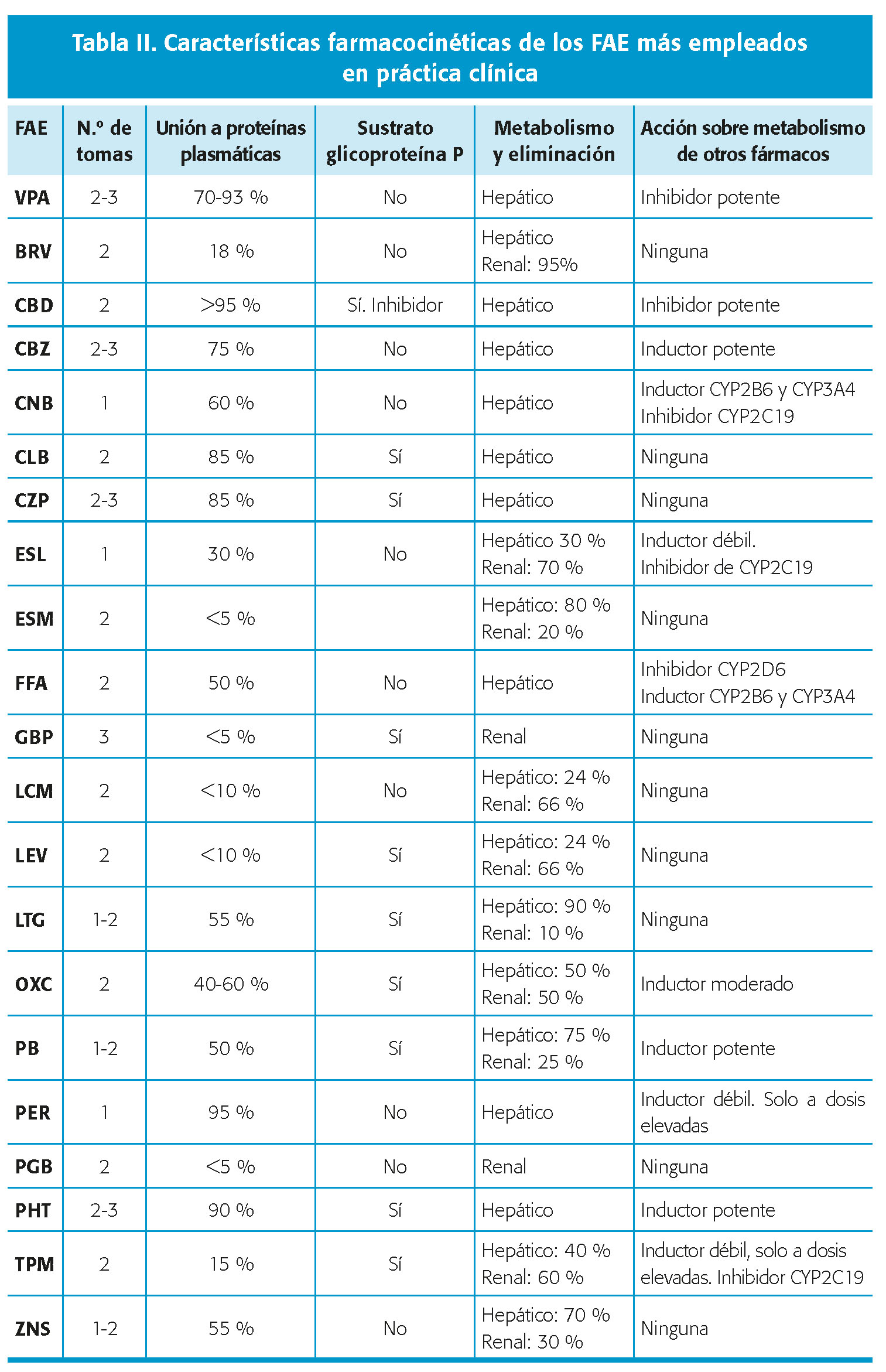
Las características farmacocinéticas de los FAC más utilizados en práctica clínica se resumen en la tabla II.
Interacciones entre FAC
Podemos diferenciar dos grandes tipos de interacciones:
- Farmacocinéticas: relacionadas con los procesos a que se ven sometidos los fármacos en su paso por el organismo, desde que se administra hasta su eliminación.
- Farmacodinámicas: relacionadas con su actividad biológica, mediatizada por el mecanismo de acción.
Interacciones farmacocinéticas
- Absorción: la mayoría de los FAC se absorben bien por vía digestiva. Tan sólo la GBP tiene una absorción saturable, de modo que, por encima de 1.200 mg/día, su absorción se reduce en un 10-35 %. La presencia de comida retrasa la absorción de muchos FAC, pero no modifica la cantidad total de fármaco absorbida. En consecuencia, el pico de dosis es menor, pero no cambia el área bajo la curva de concentración plasmática. De esta manera, la administración conjunta con comida permite disminuir los efectos secundarios asociados al pico de dosis sin variar la eficacia de la medicación. Los antiácidos a base de sales de aluminio y magnesio pueden alterar la absorción de algunos FAC, fundamentalmente PHT y, aún en mayor medida, la GBP3.
- Distribución: los fármacos más liposolubles tienden a circular en la sangre unidos a proteínas plasmáticas, sobre todo, la albúmina. Si coinciden varios FAC con alta afinidad por proteínas plasmáticas, pueden competir y desplazarse unos a otros, lo que aumentaría la fracción libre, a la que va ligada la eficacia y los efectos secundarios. Esto ocurre especialmente en situaciones que suelen acompañarse de hipoproteinemia, como la desnutrición o la edad avanzada.
- Transporte a través de la barrera hematoencefálica: el paso a través de barreras, tanto digestiva como hematoencefálica, está regulado por una serie de proteínas transportadoras, como la glicoproteína P, que tienen una acción detoxificante, extrayendo moléculas extrañas, como los fármacos, del sistema nervioso central. Los FAC inductores enzimáticos potentes también incrementan la expresión de estas proteínas transportadoras en los distintos epitelios, con lo que no sólo modifican el metabolismo, sino también la absorción y penetración cerebral de los fármacos que son sustratos de las mismas4.
- Metabolismo: CBZ, PHT, PB y PRM son potentes inductores de múltiples subunidades de los sistemas enzimáticos CYP450, UGT y epóxido-hidrolasa. En consecuencia, aceleran el metabolismo y disminuyen los niveles plasmáticos de todos los fármacos y sustancias orgánicas (vitaminas y hormonas) que se metabolizan por estas vías. OXC y ESL tienen una acción inductora intermedia, que se contrarresta en parte porque inhiben otras subunidades de CYP450, como CYP2C19, responsable fundamental del metabolismo de PHT. Por último, TPM y PER son inductores débiles, con una acción sólo evidente a dosis muy elevadas5.
La inducción enzimática se produce mediante cambios epigenéticos. Se instaura progresivamente y tarda unos días en alcanzar el nivel estable. El uso de FAC inductores potentes puede reducir los niveles plasmáticos y disminuir la eficacia de otros fármacos de metabolismo hepático, incluyendo a ellos mismos. Así, el incremento progresivo de dosis de CBZ no causa un aumento proporcional de sus niveles plasmáticos, debido a la autoinducción de su metabolismo. Si existe una sinergia farmacodinámica, se puede compensar en parte la reducción de eficacia provocada por la bajada de niveles plasmáticos del fármaco inducible. No obstante, en general, cuando se combina un FAC de metabolismo hepático con un inductor potente, es necesario utilizar dosis más elevadas que cuando se combina con un inductor débil o con un no inductor. También, la vida media del fármaco inducible se acorta. Esto tiene escasa relevancia para fármacos con una vida media muy larga, como PER. Sin embargo, con fármacos de vida media más corta, puede ser importante y obligar a un incremento en el número de tomas, para evitar periodos de infradosificación entre una dosis y otra. La supresión de FAC inductores lleva aparejada un incremento en la concentración, y en los efectos secundarios, provocados por otros fármacos que pueda estar tomando el paciente. Al contrario de lo que ocurre con la inducción, la inhibición es inmediata. El VPA es el inhibidor más potente. Cuando se añade un FAC de metabolismo hepático, como LTG o CBZ, a un paciente que toma VPA, es recomendable comenzar con una dosis más baja y realizar un escalado más lento, para evitar efectos indeseables. También la dosis de mantenimiento será más baja. Si se retira VPA, habrá que tener en cuenta que los niveles del otro fármaco también disminuirán al cesar la inhibición.
Interacciones farmacodinámicas
Son las que se producen en la diana sobre la que actúa el fármaco. Pueden modificar tanto la eficacia como la aparición de efectos secundarios.
Según el sentido en que se produzca la interacción, diferenciamos tres tipos:
- Sinérgica: la acción obtenida con el conjunto de los dos fármacos es superior a la suma de los efectos de cada uno de ellos. Se consigue generalmente mediante la combinación de fármacos con diferente mecanismo de acción. Es la interacción más favorable.
- Aditiva: el efecto obtenido con el conjunto de los dos fármacos equivale a la suma del efecto de cada uno de ellos.
- Antagónica: es la que se produce cuando la acción conjunta de los dos fármacos es inferior a la esperable por la suma del efecto de ambos.
La interacción farmacodinámica puede ser de diferente signo en lo que se refiere a eficacia y efectos secundarios. Lo ideal es encontrar combinaciones con sinergia en la eficacia y antagonismo en efectos adversos6. La determinación de las interacciones farmacodinámicas es compleja. La mejor conocida es la sinergia entre VPA y LTG. También se ha sugerido con la combinación de otros fármacos de diferente mecanismo de acción, como VPA y ESM para el tratamiento de las ausencias, o VPA y TPM.
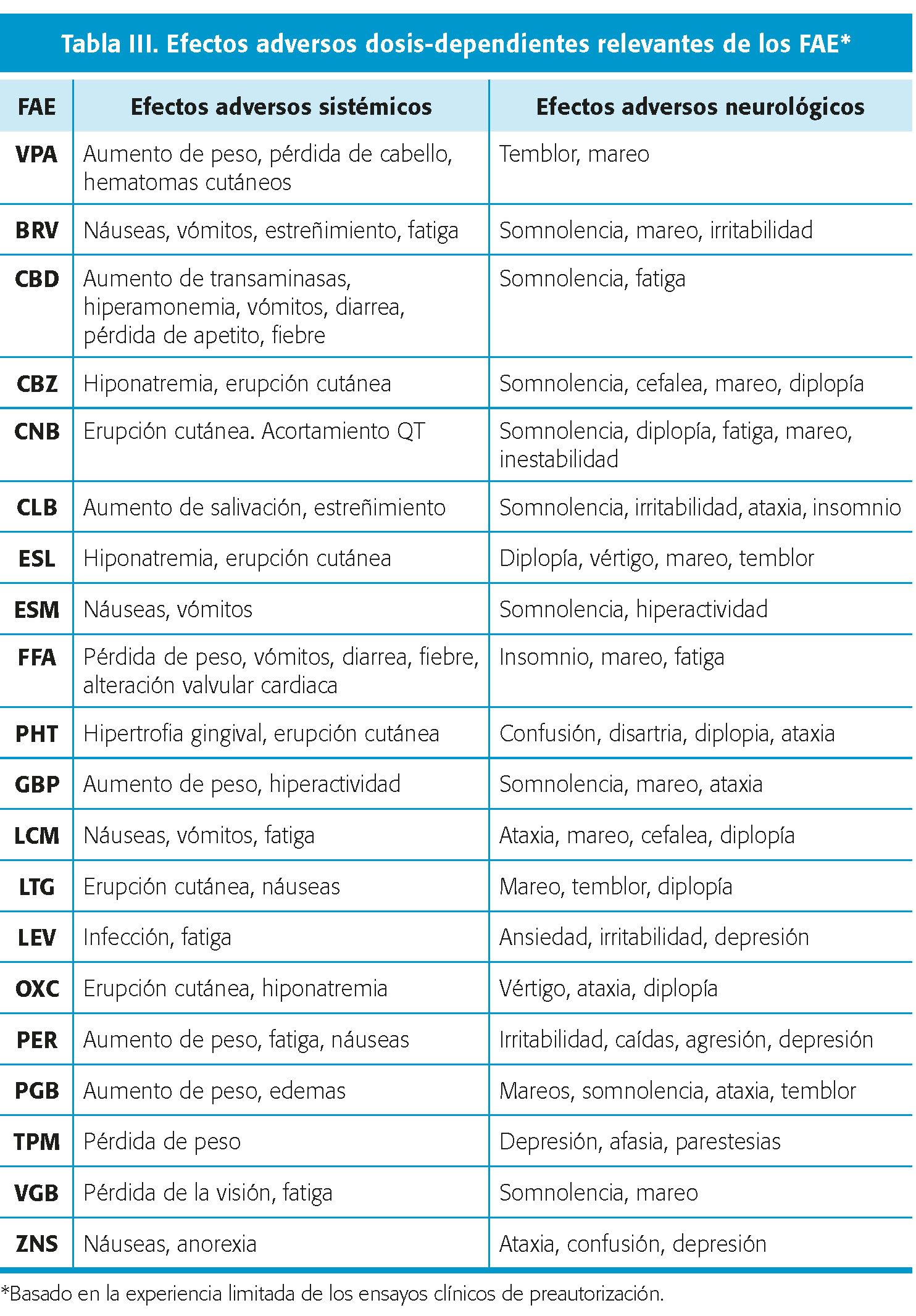
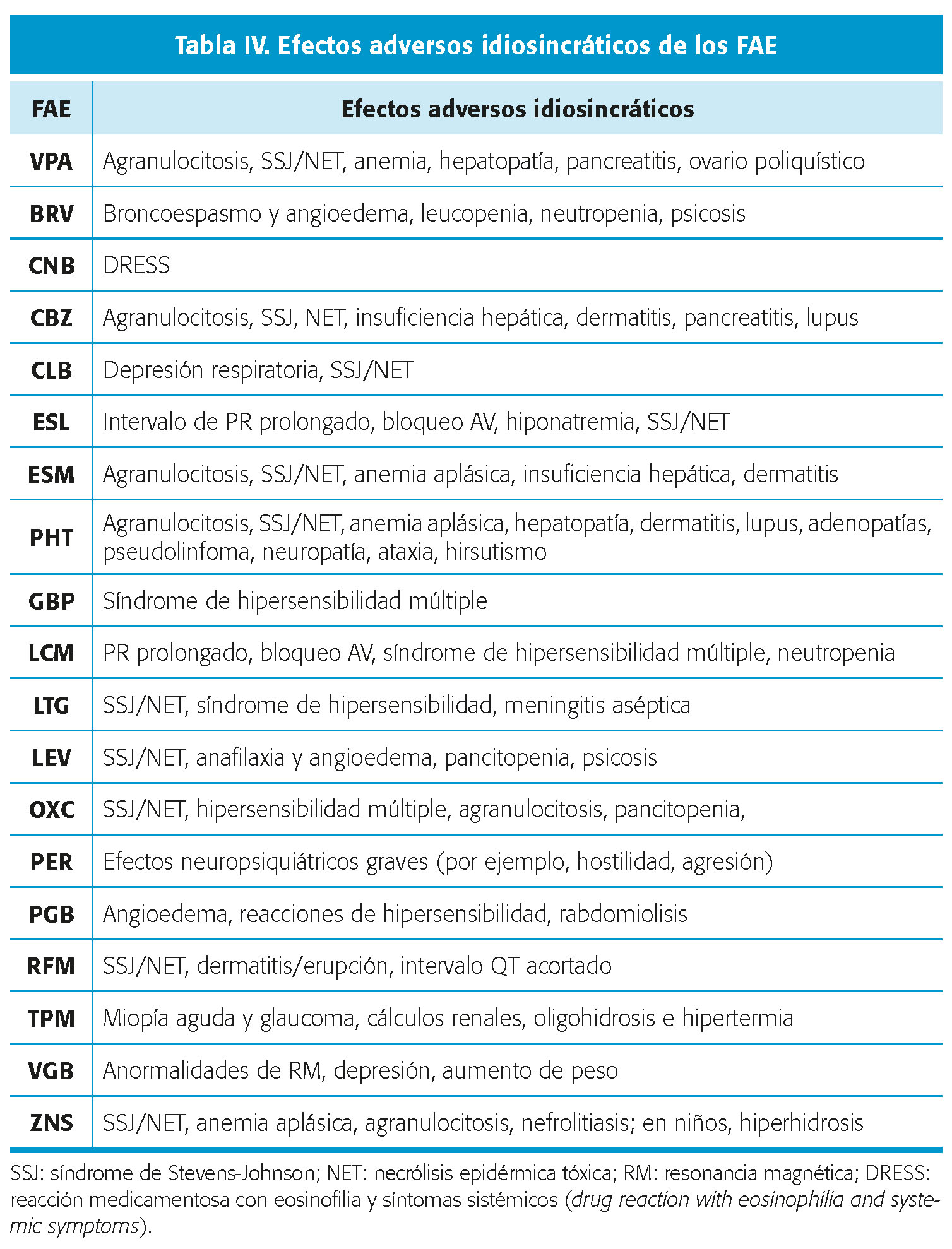
EFECTOS ADVERSOS DE LOS FÁRMACOS ANTICRISIS (TABLA III Y TABLA IV)
El impacto clínico y la frecuencia de presentación de estos efectos dependerán de las características del paciente (edad, sexo, comorbilidad asociada) y del tratamiento concomitante7,8.
- Efectos adversos dosis-dependientes (tipo A): son los más frecuentes. Suelen presentarse de forma aguda al inicio y, con el tiempo, pueden disminuir y ser tolerados. Se asocian al incremento de dosis, a la titulación rápida o a niveles plasmáticos altos. Desaparecen con la reducción de la dosis y es inhabitual que obliguen a la retirada del FAC. Pueden presentar traducción sistémica o ser neurotóxicos (tabla III).
- Efectos adversos idiosincráticos (tipo B): son menos frecuentes que los anteriores e impredecibles. Se producen por diferentes vías: reacciones de hipersensibilidad, interacción con órganos diana erróneos o por citotoxicidad directa del FAC o de sus metabolitos. Una de las presentaciones más graves es el síndrome de hipersensibilidad a los FAC. Aparece en 1/10.000 pacientes expuestos. Se caracteriza por fiebre, rash cutáneo, linfadenopatías y condicionar un fracaso multisistémico, con mortalidad del 50 %. El tratamiento consiste en la retirada del FAC y adición de corticoides e inmunoglobulinas intravenosas. (tabla IV).
- Efectos adversos crónicos (tipo C): presentan un curso insidioso, mala tolerabilidad y pueden aparecer tras exposición prolongada, son causa de retirada: efectos sobre la cognición, antiestéticos (alopecia, ganancia ponderal, hiperplasia gingival, hirsutismo e hiperpigmentación de mucosas), endocrinológicos (déficit de vitamina D, osteomalacia, disfunción sexual, hipotiroidismo), urológicos (litiasis renal) y visuales (hiperpigmentación retiniana, reducción del campo visual).
- Efectos teratógenos* y carcinogénicos (tipo D)9: los efectos carcinogénicos se han relacionado con PB y PHT en modelos animales. El tratamiento prolongado con PHT se relaciona con pseudolinfoma (simula un linfoma y se resuelve con la suspensión del FAC).
*Los efectos teratógenos se describirán en otro capítulo.

Bibliografía
- Armijo JA, Herranz Fármacos anticrisis y anticonvulsivos. En: Flórez J, Armijo JA, Mediavilla A (eds.). Farmacología Humana. 5.ª ed. Barcelona: Elsevier; 2008; p. 579-605.
- Stephen LJ, Brodie MJ. Pharmacotherapy of epilepsy: newly approved and developmental CNS Drugs. 2011; 25: 89-197.
- Yagi T, Naito T, Mino Y, Umemura K, Kawakami J. Impact of concomitant antacid administration on gabapentin plasma exposure and oral bioavailability in healthy adult Drug Metab Pharmacokinet. 2012; 27: 248-54.
- Löscher W, Schmidt Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia. 2006; 47: 1253-84.
- Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: interactions between antiepileptic drugs and other Lancet Neurol. 2003; 2: 473-81.
- Zaccara G, Perucca E. Interactions between antiepileptic drugs, and between antiepileptic drugs and other Epileptic Disord 2014; 16: 409-431
- Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002; 58(8 Suppl 5): S2-8.
- Zaccara G, Giovannelli F, Giorgi FS, Franco V, Gasparini S, Benedetto Tolerability of new antiepileptic drugs: a network meta-analysis. Eur J Clin Pharmacol. 2017; 73(7): 811-7.
- Perucca P, Gilliam FG. Adverse effects of antiepileptic drugs. Lancet Neurol. 2012; 11(9): 792-802. Erratum in: Lancet 2012; 11(9): 746.
