
INTRODUCCIÓN
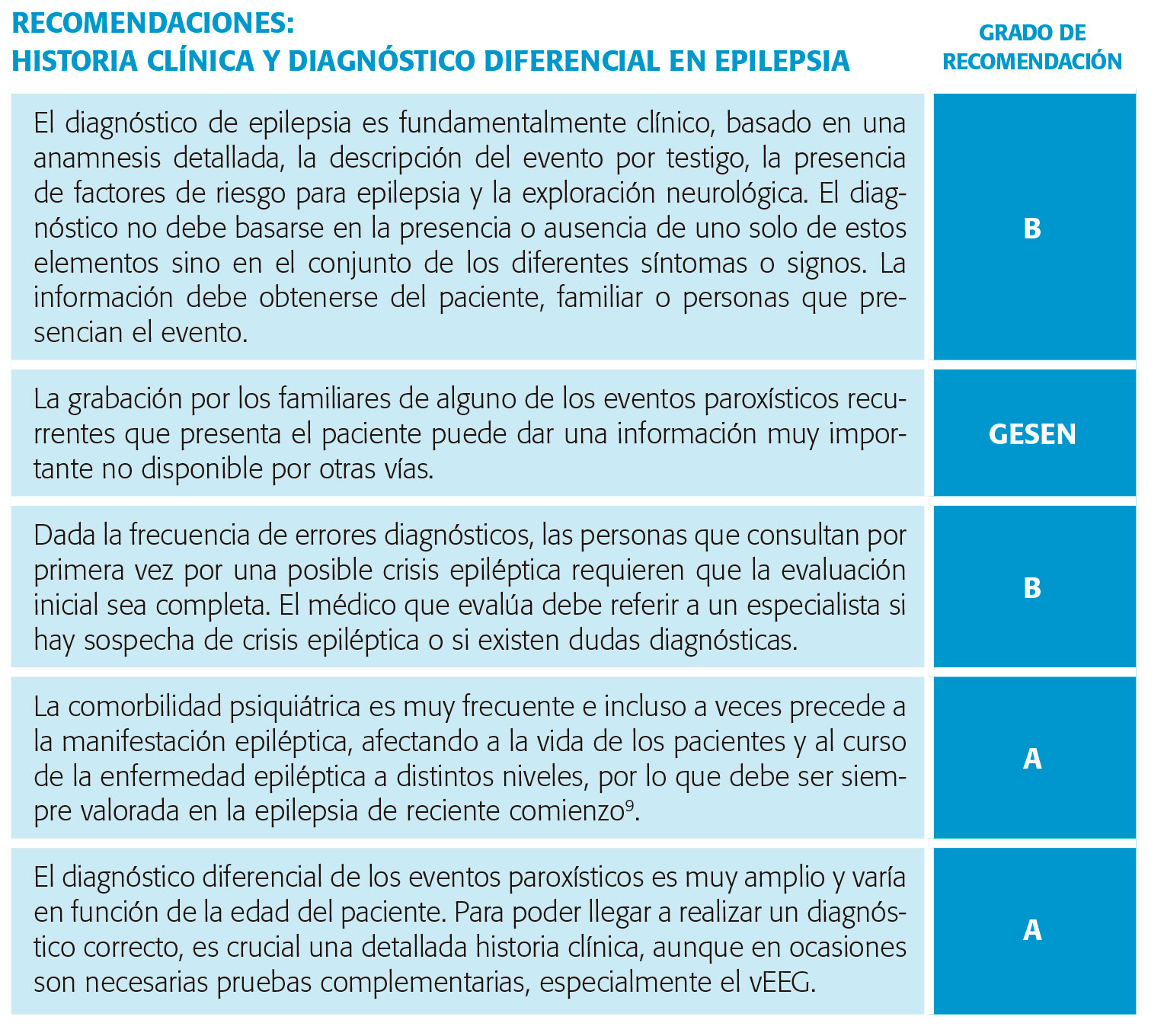
Aicardi y Taylor refieren que el diagnóstico clínico es un proceso intelectual mediante el cual todas las posibles fuentes de información, desde la pura clínica hasta la alta tecnología, son integradas para alcanzar una conclusión magnífica5. La realización de una cuidadosa y detallada historia clínica es el paso inicial del adecuado diagnóstico en epilepsia. “El diagnóstico será tan bueno como lo sea la historia clínica”.
Recientemente Nowacki y Jirsch publican en la revista Seizure (2017)6 una revisión de la literatura sobre los puntos más importantes de la historia clínica y el examen físico en la evaluación de la primera crisis, afirmando que aún en la era de los regis tros EEG digitales y la neuroimagen, la evaluación clínica inicial sigue siendo esencial para el diagnóstico, tratamiento y pronóstico de los pacientes que sufren una prime ra crisis epiléptica. Nivel de evidencia II.
ANAMNESIS (TABLA V)
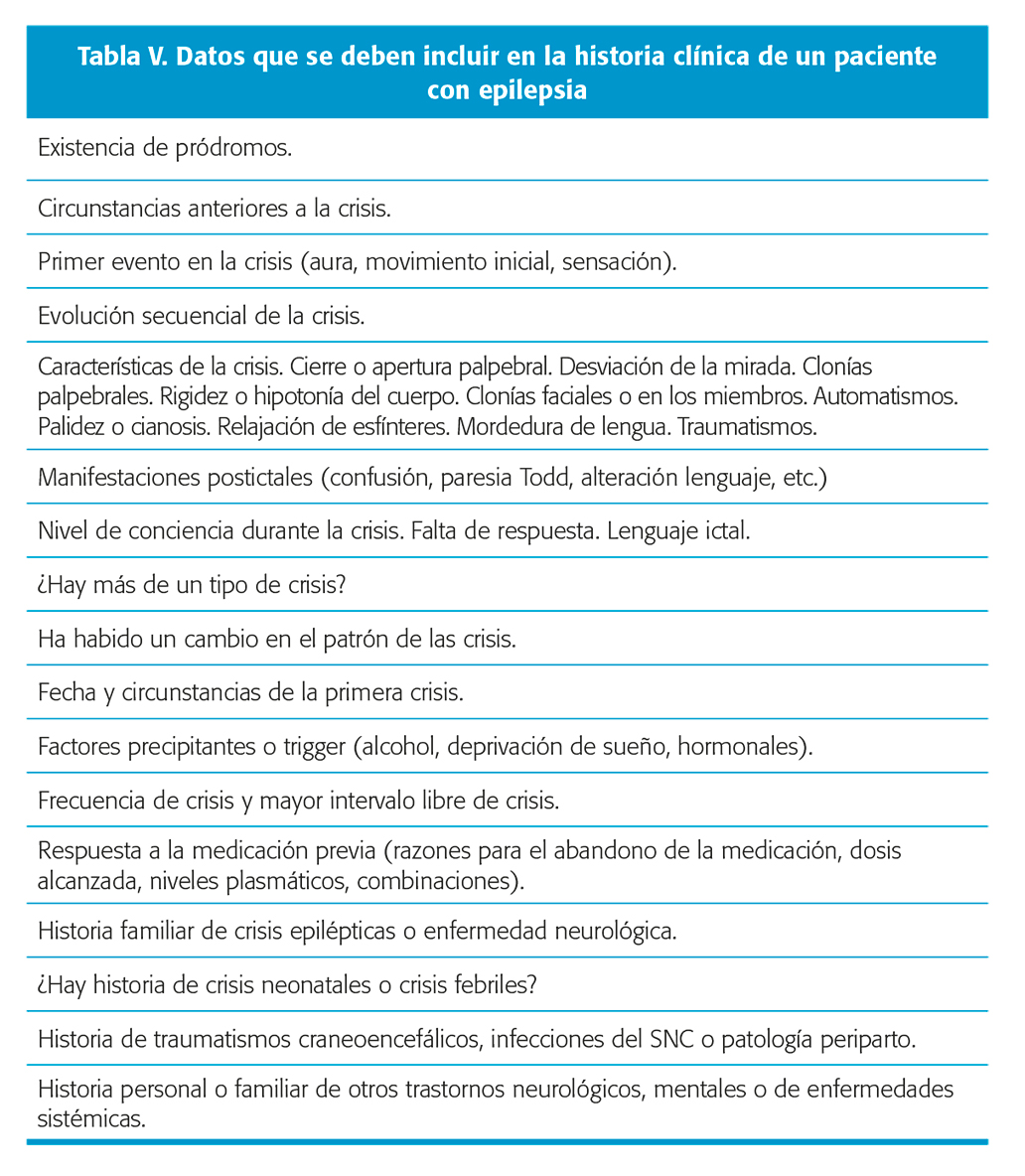
La aproximación inicial al paciente con epilepsia se hace en gran parte a través de la anamnesis, especialmente por una adecuada descripción del evento ocurrido. La información debe obtenerse del paciente cuando es posible, así como de otras personas, sobre todo familiares, que puedan haber presenciado los ataques típicos. La memoria es altamente selectiva, y la brusquedad e imprevisión de las crisis, especialmente la primera, puede hacer que el relato de la misma sea inconsistente. También los recuerdos del paciente pueden estar limitados por la alteración de la conciencia y la amnesia postictal. La estimación de la duración de la crisis es casi invariablemente exagerada5. Nivel de evidencia III.
La anamnesis del enfermo con epilepsia debe satisfacer unos requisitos básicos, si bien la revisión de la literatura no da apoyo definitivo a un determinado modelo de historia clínica, por lo que las evidencias en este aspecto emanan de las opiniones de expertos. Nivel de evidencia IV. Obtener una descripción del evento paroxístico es la parte más importante de la realización de la historia clínica y requiere un alto grado de atención y considerable tiempo. Hacer la historia de una alteración paroxística necesita una técnica propia de interrogatorio médico y un buen conocimiento de las manifestaciones complejas de la epilepsia. A menudo es útil y siempre recomendado preguntar por la descripción de un ataque específico. El último suele ser el mejor recordado. Una descripción de la primera crisis es de gran interés ya que no infrecuentemente son diferentes a las subsecuentes.
Los datos fundamentales que deben recabarse son: la pregunta de si existió pérdida de conocimiento y si fue completa o desconexión parcial. Se debe preguntar por pródromos que indican que puede venir una crisis: cambios de comportamiento, inestabilidad, somnolencia, sensación de miedo, salivación, hipotermia, etc.… La investigación de un estímulo regularmente asociado al evento es de interés y debe ser sistemáticamente preguntado: trauma menor o sangre (síncope), luces intermitentes, sobresalto. El análisis de las auras es importante por su posible valor localizador. Nivel de evidencia III.
Se debe preguntar por la presencia de movimientos anómalos, su tipo y secuencia de aparición (versión cefálica, automatismos orales o de extremidades, rigidez, clonías, mioclonías) y si éstos se inician focalmente o son generalizados. La reconstrucción de la secuencia ictal es difícil, muchas crisis son breves y rápidamente cambian de síntomas. Es esencial valorar el nivel de conciencia, la responsividad, la memoria de los eventos, la alteración cognitiva o del lenguaje, incontinencia o mordedura de lengua. Algunos datos semiológicos también han demostrado tener un valor pronóstico. En concreto en el caso de la epilepsia temporal la consignación de la presencia de actividad automática crítica aislada tiene valor predictivo7, siendo del 62,3 % para el desarrollo de EFR. Nivel de evidencia III. Las circunstancias de ocurrencia, duración y posible presencia de fenómenos postictales, tales como paresia de Todd o afasia tienen indiscutible valor para lateralización y localización. El lugar o ambiente en el cual se producen los ataques puede tener un considerable significado (crisis no epilépticas). Puntos de particular interés: relación con el ciclo vigilia-sueño (si está despierto, durmiendo, al principio del sueño, antes o después); cuando ocurren (mañana, tarde, noche); tipo de actividad (descanso, ejercicio, cama, baño, comiendo, actividad intelectual, usando ordenador o viendo televisión, alteración emocional); estado de salud (enfermedad sistémica o fiebre reciente).
Debemos también interrogar sobre episodios previos similares o por cuadros de desconexión del medio, auras o movimientos involuntarios (mioclonías, clonías). No es infrecuente que en una supuesta crisis tónico-clónica descubramos que hubo eventos previos como ausencias breves, mioclonías o clonías focales que han pasado inadvertidos o por los que el paciente no ha consultado y que constituían manifestaciones previas más leves de un síndrome epiléptico (esto sucede con relativa frecuencia en algunas epilepsias generalizadas como la epilepsia de ausencias infantil o juvenil y la epilepsia mioclónica juvenil).
El diagnóstico de epilepsia y crisis epilépticas es clínico. Es muy importante lograr un diagnóstico preciso del tipo de crisis que, con ayuda de estudios complementarios, permitirá determinar el síndrome epiléptico y escoger el mejor tratamiento. Nivel de evidencia IV.
Familiares de pacientes que tienen episodios recurrentes pueden intentar grabar alguno, lo que da una información muy importante que no se puede obtener por otras vías.
La historia, sin embargo, no está limitada a esta descripción, hay que obtener la historia del problema, pero también la historia de la persona, ya que las personas reaccionan de su propia forma en un determinado lugar. Hay que valorar el impacto en la vida del paciente, desde el punto de vista físico, pero también psicosocial (enfermedad crónica).
Antecedentes personales
- Antecedentes obstétricos: patología durante el embarazo, distocias del parto y sufrimiento fetal, realizando la anamnesis sobre familiares con capacidad para
- Hitos del desarrollo psicomotor y rendimiento
- Existencia o no de convulsiones febriles durante la
- Antecedentes de TCE en la infancia o infecciones del sistema nervioso central (SNC).
- Comorbilidades, con especial referencia a comorbilidad psiquiátrica, neurológica y afectaciones somáticas de hígado o riñón.
- Historial de fármacos antiepiléptico (FAE) previos y Esta información es de especial relevancia para clasificar el grado de refractariedad de la epilepsia según los criterios de la ILAE.
- Es importante también el consumo de alcohol y otros tóxicos, así como otros medicamentos de consumo reciente o actual, especialmente psicótropos.
Antecedentes familiares
Preguntar con especial atención a los de epilepsia o enfermedades neurológicas en la familia. Es importante preguntar sobre antecedentes familiares de crisis epilépticas, crisis febriles, síncopes, cardiopatías, nefropatías y enfermedades psiquiátricas.
EXPLORACIÓN FÍSICA
El objetivo del examen físico es buscar cualquier evidencia de la causa subyacente, si está limitada al cerebro o afecta otros sistemas, como es el caso de alteraciones neurocutáneas, anormalidades cromosómicas y algunas enfermedades sistémicas.
La exploración sistemática debe incluir de manera ineludible las siguientes actividades6:
- Inspección general: con un examen de la piel y de la constitución general, puesto que la epilepsia puede formar parte de numerosos síndromes cutáneos y dismórficos.
- Exploración cardiovascular: medición de la tensión arterial, anomalías del ritmo cardiaco o soplos cardiovasculares. Esta exploración resulta de especial interés en casos de diagnóstico diferencial con eventos de origen cardiogénico.
- La exploración neurológica en el paciente epiléptico, por su parte, debe tener el objetivo principal de poner en evidencia, en caso de que existan, signos indicativos de hipertensión intracraneal, disfunción neurológica focal, meníngea o en la esfera cognitiva, que orienten el diagnóstico a una epilepsia sintomática. Nivel de evidencia IV. Anotar el intervalo desde la última
- Valoración cognitiva y examen del estado psíquico del paciente: es fundamental para la detección del principal diagnóstico diferencial de las crisis epilépticas (CE), como las pseudocrisis de origen psicógeno.
El diagnóstico incorrecto de epilepsia es frecuente, alcanzando hasta el 26 % de los pacientes remitidos a una Unidad de Epilepsia para evaluación quirúrgica. Las causas de error son sobre todo fallos en la anamnesis y en segundo lugar mala interpretación del registro electroencefalográfico. Con esto el retraso diagnóstico a veces llega hasta 7 o 10 años8. Nivel de evidencia II.
Los estudios que relacionan el diagnóstico de epilepsia o la interpretación de datos semiológicos con la formación en epilepsia del médico que los realiza, observan que la tasa de errores diagnósticos es menor, llegando a un 5 % en neurólogos y neuropediatras, especialmente si son expertos en epilepsia. Nivel de evidencia III.
DIAGNÓSTICO DIFERENCIAL DE LA EPILEPSIA EN LAS DISTINTAS EDADES
En un gran número de casos, ante un evento paroxístico, una historia clínica detallada puede ser suficiente para poder hacer un diagnóstico correcto, aunque a veces es necesario el uso de pruebas complementarias (principalmente, vídeo-electroencefalograma (vEEG), aunque también puede ser necesario: electrocardiograma, test de la mesa basculante, etc.).
En función de la edad del paciente, se han descrito diferentes eventos paroxísticos no epilépticos9. Nivel evidencia II.
Eventos paroxísticos no epilépticos en el neonato
En esta franja de edad, se pueden observar movimientos, gestos y actitudes relacionados con la maduración cerebral10. Nivel evidencia II.
Temblores
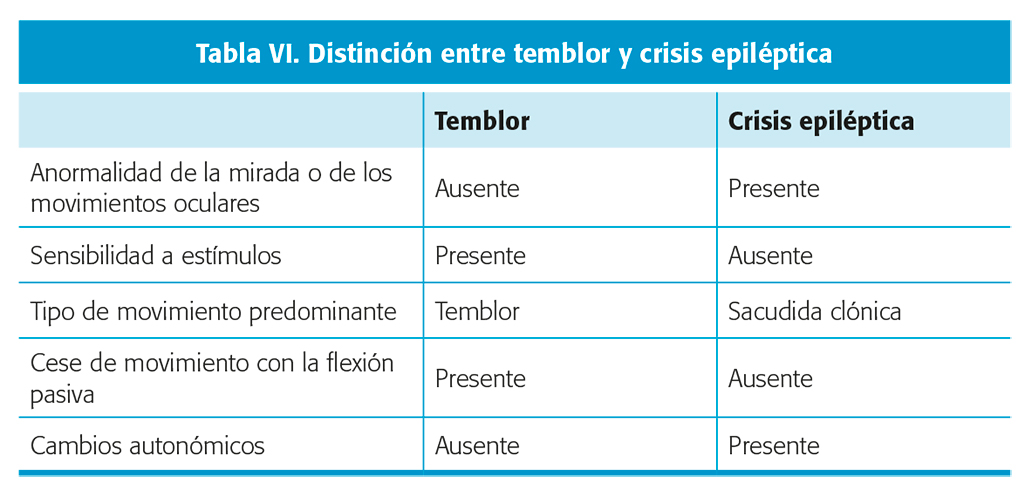
Este fenómeno se observa de forma frecuente en los neonatos. Se han descrito dos tipos: un temblor “fino”, asociado frecuentemente con el llanto, y que se define por una alta frecuencia (seis o más veces por segundo) y una baja amplitud (menor a 3 cm). Estos temblores suelen acompañarse de hiperactividad, bajo umbral para las respuestas reflejas y reflejo de Moro de fácil aparición, constituyendo el síndrome de hiperexcitabilidad neonatal. Por otro lado, encontramos los temblores “intensos (menos de 6 veces por segundo y amplitud mayor a 3 cm), que suelen estar asociados a trastornos metabólicos, problemas sistémicos o enfermedades neurológicas. Los temblores se suelen resolver en un periodo promedio de 7.2 meses, pudiendo ser mayor si el temblor se acompaña de hiperexcitabilidad. Las principales diferencias entre temblor y crisis epiléptica se explican en la tabla VI.
Mioclonías benignas del sueño
Habitualmente, se inician en neonatos menores de dos semanas, y suelen desaparecer, en el 95 % de los casos, antes de los 6 meses de edad. Las mioclonías preferentemente suceden en el sueño sin movimientos oculares rápidos (NREM), en series de 20 a 30 por minuto, y duran de promedio menos de una hora. Raramente se observan en la cara, cabeza y abdomen.
Hiperekplexia
Se define como una exagerada respuesta ante un estímulo inesperado. Puede ocurrir una apnea central (disfunción troncal) o periférica (laringoespasmo, aspiración o bloqueo cardiaco). Puede recurrir en la adolescencia ante estímulos súbitos, frío o gestación.
Eventos paroxísticos no epilépticos en el lactante
Relacionados con el movimiento11. Nivel de evidencia II
- Spasmus nutans: consisten en episodios de nistagmo asimétrico asociado a inclinación y movimientos de afirmación o negación de la cabeza. Desaparecen alrededor de los 6 años.
- Estremecimientos: se caracterizan por la detención de la actividad sin pérdida de consciencia, temblor brusco rápido de corta duración, especialmente en la cabeza con posturas tónicas con flexión o extensión de la cabeza y cuello con estremecimiento cefálico y de Suelen iniciarse a los 4 meses de edad.
- Desviación de la mirada hacia arriba con ataxia: suelen ser episodios prolongados, que se inician entre los 3 meses y los 2 años. Remite espontáneamente en unos años, aunque la mitad de los casos evoluciona con un retraso del desarrollo y del
- Vértigo paroxístico benigno: episodios breves de inestabilidad sin aura, asociados a sensación de miedo y clínica autonómica. Se inician entre los 2 y 4 años, y remiten hacia los 5 años. Se relacionan con migraña.
- Síndrome de Sandifer: se caracteriza por posturas anómalas del cuello, tronco y extremidades, pudiendo presentar episodios súbitos de tortícolis, rigidez generalizada y opistótonos, junto con apnea, mirada fija y mioclonías de miembros, como consecuencia de un reflujo gastroesofágico. Es característico que suceda después de Especialmente se presenta entre los 18-36 meses.
- Mioclonías benignas del lactante: suelen presentarse al despertar en forma de salvas repetidas en forma de flexión de cuello, y extensión y abducción de los miembros Suelen iniciarse a los 3 meses, y desaparecer hacia los 9 meses.
- Crisis tónicas reflejas del lactante: consisten en una contracción tónica con extensión de las 4 extremidades, apnea y cianosis, con congestión facial durante 3-10 Se desencadenan por estímulos táctiles o cambios posicionales. Suele presentarse a los 2-3 meses, y desaparecen espontáneamente pocos meses después.
- Estereotipias: conductas motoras repetitivas que se presentan de forma rítmica y con Suelen iniciarse al año y ceder a los 2-3 años, aunque son más acentuadas y persistentes en pacientes con enfermedad neurológica (por ejemplo, autismo).
- Body rocking: se caracteriza por movimientos de balanceo, estando en sedestación, con movimientos rítmicos de tronco y cabeza tanto en dirección lateral como postero-anterior. Se inician en el primer año de vida, siendo más frecuentes en lactantes poco estimulados, con discapacidad o déficit
Relacionados con la hipoxia
- Espasmos del sollozo: existen 2 tipos: el pálido, que corresponde a un síncope vaso-vagal por un mecanismo cardioinhibitorio neurológicamente mediado; y el cianosante, en contexto de una apnea espiratoria prolongada, que se acompaña de pérdida de consciencia, cianosis, y en ocasiones posturas tónicas. Suelen aparecer entre los 6-18 meses, y ser desencadenados, además del llanto, por pequeños traumatismos, miedo o frustración.
Relacionados con el sueño
- Episodios de apnea: se caracterizan por el cese de la respiración en sueño durante más de 12-15 Suelen observarse en los primeros meses de vida en niños prematuros, y pueden ser secundarios a inmadurez, reflujo gastroesofágico o medicación. Se aconseja monitorización continua durante los meses posteriores al evento, dada su relación con muerte súbita.
- Head banging: consiste en movimientos rítmicos de la cabeza cuando el lactante va a Suelen remitir a los 2-3 años, y son más frecuentes e intensos en niños con discapacidad neurológica o sensorial.
Relacionados con el dolor
- Vómitos cíclicos del lactante: se definen como episodios agudos y repetitivos de náuseas y vómitos. Se repiten con cierta periodicidad (3-4 por año) y pueden durar varios días. Se ha descrito la aparición de migraña en edades
- Dolor paroxístico extremo: suele manifestarse a nivel rectal y en región perianal, junto con rubefacción facial, y suele acompañarse de rigidez y espasmo tónico, que en el contexto de bradicardia y asistolia, puede finalizar en un cuadro
Miscelánea
- Torticolis paroxístico: suele cambiar de un lado a otro en cada episodio, y puede asociarse a malestar, irritabilidad, agitación, palidez, vómitos o incluso Comienza en el primer año de vida y desaparece antes de los 3-5 años. Se ha relacionado con la migraña.
- Episodios de autoestimulación: suceden al estimular los genitales directamente o a través de maniobras de frotamiento de los muslos, desencadenando posturas distónicas, gruñidos, diaforesis, agitación, cianosis o palidez, mirada perdida o contorsión pélvica. Se observan entre los 3 meses y los 5 años.
- Hemiplejía alternante de la infancia: se caracteriza por ser una hemiplejía flácida subaguda, junto con clínica autonómica y nistagmo monocular ipsilateral. Se puede iniciar con posturas tónicas o distónicas, junto con agitación y sensación de miedo. Puede durar de pocos minutos a varias horas. Se recupera completamente tras el sueño. No debuta antes de los 18 meses, y los episodios tienden a remitir con la
Eventos paroxísticos no epilépticos en la infancia12
- Tics: los tics son movimientos o sonidos repetitivos, estereotipados, repentinos y no El estrés los empeora, y desaparecen al dormir. A destacar que hasta un 25 % de los niños escolares presentan tics. Aunque en la mayoría, los tics desaparecen en menos de un año.
- Trastornos hipercinéticos:
– Discinesia paroxística cinesigénica: se caracteriza por movimientos breves, generalmente coreiformes o de atetosis, que pueden ser hemicorporales o bilaterales, provocados por movimientos súbitos, como levantarse de una silla.
- Discinesia paroxística no cinesigénica: en estos casos el factor desencadenante es el estrés, el alcohol o la cafeína.
- Distonía paroxística inducida por el ejercicio: se manifiesta tras varios minutos después de terminar el
- Mirada fija y paro del comportamiento: se caracterizan por una detención de la actividad, con una expresión facial indiferente y mirada fija sin Suelen ocurrir si el niño está aburrido o inactivo, y terminan al usar estímulos táctiles o auditivos de elevada intensidad.
- Parasomnias:
- Del sueño NREM: ocurren una o dos horas después de iniciar el sueño, se caracterizan por ser episodios recurrentes de medio despertar, con falta de respuesta a estímulos, y amnesia de los Son comunes en niños de 3 a 13 años, y generalmente desaparecen en la adolescencia.
- Terrores nocturnos: es la parasomnia NREM más frecuente en la infancia. Se caracteriza por un grito agudo y conmovedor, asociado a cambios autonómicos (sudoración y taquicardia) con cambios del comportamiento, como miedo intenso de duración prolongada, con dificultad para despertar al
- Despertar confusional: los niños despiertan parcialmente, aparentemente confundidos, sin responder a estímulos, pese a que parecen que están despiertos.
- Sonambulismo: el paciente despierta parcialmente del sueño NREM y puede realizar diferentes En niños, la prevalencia es de alrededor de un 7 %.
- Del sueño REM: las más comunes son las pesadillas, que pueden estar causadas por fármacos, como los antidepresivos, narcóticos y barbitúricos. Otras menos frecuentes son: enuresis, emesis cíclica, temblores, y ataques de
- Del sueño NREM: ocurren una o dos horas después de iniciar el sueño, se caracterizan por ser episodios recurrentes de medio despertar, con falta de respuesta a estímulos, y amnesia de los Son comunes en niños de 3 a 13 años, y generalmente desaparecen en la adolescencia.
Eventos paroxísticos no epilépticos en la adolescencia
Crisis psicógenas: son el evento paroxístico no epiléptico más frecuente en adolescentes.
Eventos paroxísticos no epilépticos en adultos13
- Síncope. Se caracteriza por una perdida transitoria de consciencia y del tono postural, en contexto de una abrupta reducción del flujo sanguíneo cerebral. Es característico que inicialmente el paciente presente una serie de síntomas (pródromos) que incluyen de visión borrosa a pérdida de la visión, tinnitus, y sensación de mareo. A los que también pueden añadirse síntomas autonómicos como palidez, rubor, sudoración fría, sensación de calor, náuseas y molestias La rigidez y los movimientos tónico-clónicos pueden llegar a ocurrir, y son de breve duración, típicamente no rítmicos. Es característico un muy breve
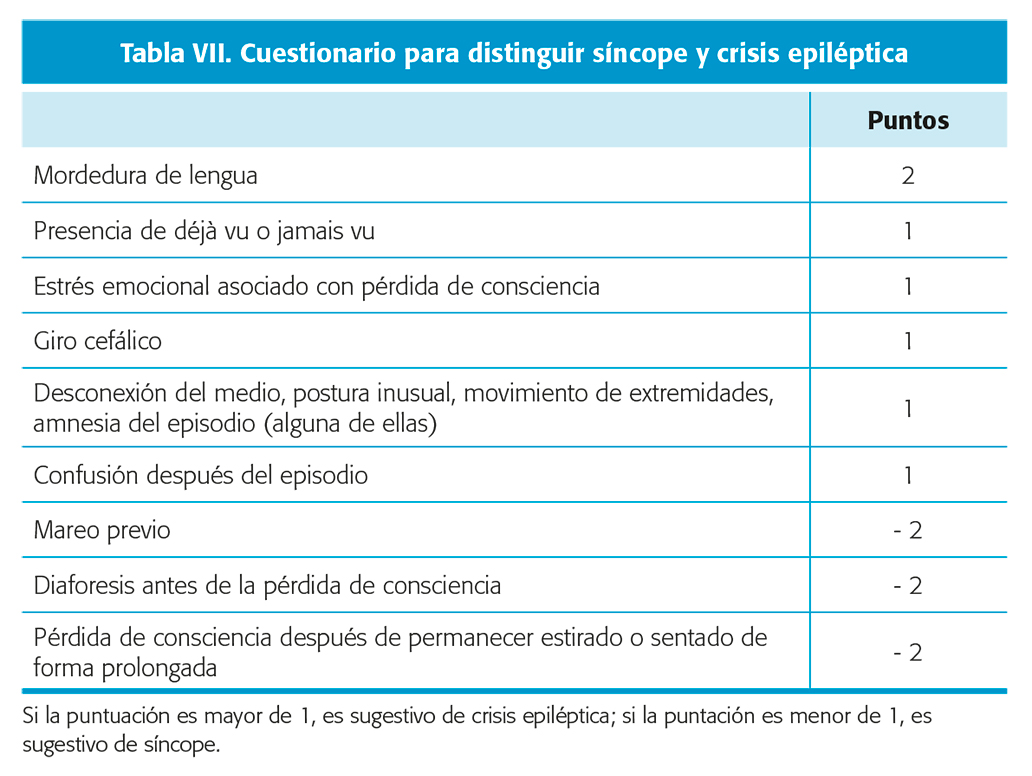 o inexistente estado confusional postepisodio. Por otro lado, la relajación del esfínter vesical es bastante frecuente. No así la mordedura de lengua (aunque típicamente en la punta de la misma, si es lateral sugiere una crisis epiléptica). Se ha diseñado un cuestionario para intentar distinguirlos de las crisis epilépticas (tabla VII) 14. Nivel de evidencia III.
o inexistente estado confusional postepisodio. Por otro lado, la relajación del esfínter vesical es bastante frecuente. No así la mordedura de lengua (aunque típicamente en la punta de la misma, si es lateral sugiere una crisis epiléptica). Se ha diseñado un cuestionario para intentar distinguirlos de las crisis epilépticas (tabla VII) 14. Nivel de evidencia III.
En función de la causa del síncope, se distinguen diferentes tipos:
- Síncope vaso-vagal (neuro-mediado): es causado por una respuesta desproporcionada de los reflejos Los típicos factores desencadenantes son: bipedestación prolongada, deshidratación (lugares calurosos o congestionados de gente), cambios posturales bruscos (de decúbito a bipedestación), dolor o emociones intensas. Es común que haya historia familiar de síncopes vaso-vagales.
Una variante de este síncope es el síncope del seno carotídeo, en el que se manifiesta la clínica anteriormente descrita en el contexto de manipulación mecánica accidental del seno carotídeo. Otro tipo es el síncope situacional, que está asociado a situaciones específicas (micción, toser, defecar, etc.).
- Síncope ortostático: se produce como consecuencia de cambios posturales. Puede ser provocado por fármacos hipotensores, como por neuropatías periféricas en contexto de diabetes o
- Síncope cardiaco: es el resultado de alteraciones en el ritmo cardiaco o de anomalías estructurales cardiacas. Debe sospecharse ante historia de síncopes en sueño, y antecedentes familiares de síncope y muerte súbita.
- Trastornos del sueño.
- Cataplejía: consiste en una pérdida brusca del tono muscular, en contexto de un estímulo emocional. La presencia de otros síntomas de narcolepsia: excesiva somnolencia diurna, parálisis de sueño, o alucinaciones hipnagógicas, ayuda a hacer un diagnóstico
- Movimientos periódicos de las piernas: se definen como movimientos repetitivos y estereotipados, predominantemente durante el sueño NREM. Pueden ocurrir uni o bilateralmente, en intervalos de 20 a 40
- Parasomnias asociadas a sueño NREM: descritos en el capítulo de la
- Parasomnias asociadas a sueño REM: se caracterizan por la pérdida de atonía de la musculatura durante el sueño. Los pacientes presentan diferentes comportamientos motores (hablar, gritar, o golpear) en respuesta al contenido de sus sueños. Los pacientes no están conscientes durante los episodios, pero pueden ser despertados, y frecuentemente recuerdan el contenido de los sueños. Suelen ocurrir durante la segunda parte de la noche, y pueden durar de minutos a
- Trastornos del movimiento. Puede ser difícil distinguir entre una crisis epiléptica y un trastorno del movimiento (distonía paroxística, algunos tipos de temblor, tics o espasmos hemifaciales), por lo que el diagnóstico habitualmente estará apoyado en un
- Ictus. Los ictus isquémicos transitorios causan síntomas y signos neurológicos de breve duración que pueden confundirse con crisis epilépticas. Por otro lado, la estenosis grave de carótida puede producir un fenómeno conocido como “limbshaking”, que consiste en sacudidas clónicas rítmicas o arrítmicas en la mano, brazo y/o pierna contralateral, aunque nunca se observa una marcha jacksoniana y la cara nunca está
- Amnesia global transitoria. Se caracteriza por un inicio súbito con marcada alteración de la memoria anterógrada, desorientación temporal, y ocasionalmente desorientación en espacio, pero siempre está preservada la orientación en persona y el reconocimiento de los demás. Este episodio puede durar horas (aunque menos de 24 h), y debe ser diferenciado de un estatus epiléptico no
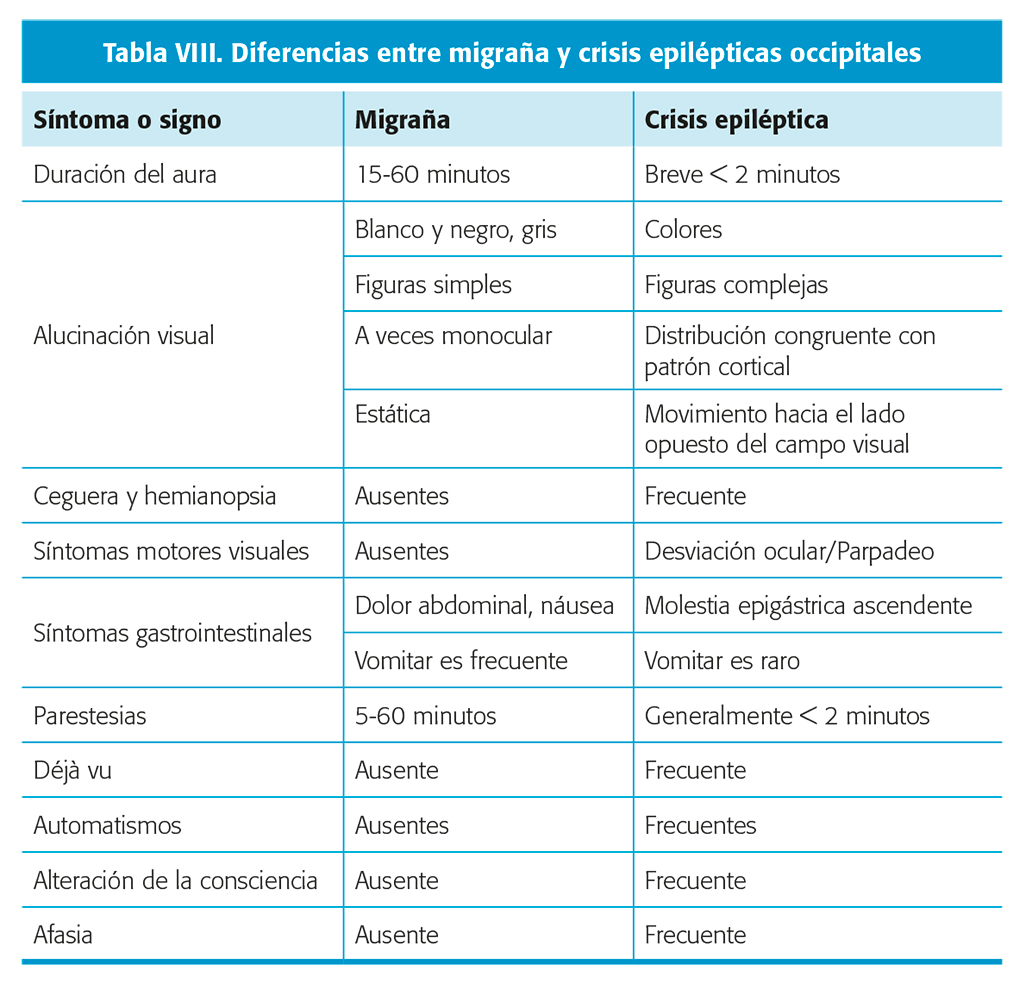
- Migraña. Las auras de la epilepsia focal occipital pueden confundirse con las auras visuales migrañosas, aunque presentan varias diferencias (tabla VIII)15.
Nivel de evidencia III.
Otro tipo de migraña que también se puede confundir con epilepsia, es la migraña basilar, que puede causar confusión y pérdida de consciencia.
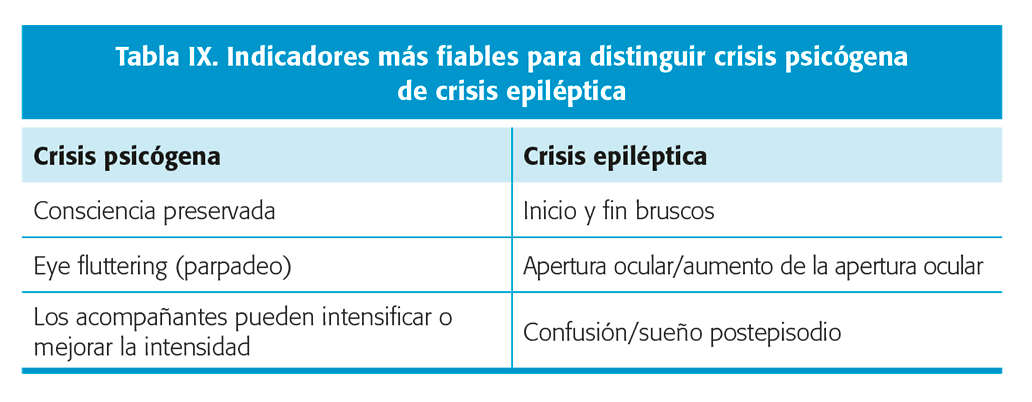
- Crisis psicógenas. Hasta un 30 % de pacientes referidos para monitorización vEEG prolongada en un centro terciario han sido diagnosticados de crisis psicógenas. El promedio de tiempo para llegar a un correcto diagnóstico es de hasta 7.2 años. Este retraso diagnóstico está asociado a un peor pronóstico. Algunos aspectos clínicos pueden ayudar a diferenciar entre crisis psicógenas y epilépticas (tabla IX)16,17. Nivel de evidencia II
Bibliografía
- Aicardi J, Taylor History and physical examination. Chapter 70. In Engel J and Pedley TA. Epilepsy: a compresive textbook. Filadelfia: Lippincott Williams and Wilkins; 2008.
- Nowacki TA, Jirsch Evaluation of the first seizure patient: key points in the history and physical examination. Seizure. 2017; 49: 54-63.
- Serrano P, Payan M, Quiroga P, Fernandez J, Parrón T. Predictive model for refractoriness in temporal lobe epilepsy base don clinical and diagnostic test data. Epilepsy 2012; 101: 113-21.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist QJMED. 1999; 92: 15-23.
- Kanner Psychiatric comorbidities in new onset epilepsy: should they be always investigated? Seizure. 2017; 49: 79-82.
- Cross JH. Differential diagnosis of epileptic seizures in infancy including the neonatal Semin Fetal Neonatal Med. 2013; 18(4): 192-5.
- García-Alix A G-AJ, Quero Evaluación neurológica del recién nacido. Madrid: Díaz de Santos; 2012.
- Fernandez-Alvarez Transient benign paroxysmal movement disorders in infancy. Eur J Paediatr Neurol. 2018; 22(2): 230-7.
- Campistol J. Trastornos paroxísticos no epilépticos en la infancia. Barcelona: Viguera Editores;
- Carreño Recognition of nonepileptic events. Semin Neurol. 2008; 28(3): 297-304.
- Sheldon R, Rose S, Ritchie D, Connolly SJ, Koshman ML, Lee MA, et al. Historical criteria that distinguish syncope from J Am Coll Cardiol. 2002; 40(1): 142-8.
- Falip M, Gil-Nagel A, Viteri Torres C, Gómez-Alonso Diagnostic problems in the initial assessment of epilepsy. Neurologist. 2007; 13(6 Suppl 1): S2-S10.
- Syed TU, LaFrance WC Jr, Kahriman ES, Hasan SN, Rajasekaran V, Gulati D, et al. Can semiology predict psychogenic nonepileptic seizures? A prospective study. Ann Neu 2011; 69(6): 997-1004.
